|
To Print: Click your browser's PRINT button.
NOTE: To view the article with Web enhancements, go to: http://www.medscape.com/viewarticle/549765 MedGenMed HIV/AIDS — Case Report Case Files From Stanford University Medical Center: Ten Years of HAART: A Long Wait for Full HIV Suppression Nancy Shulman, MD; Robert W. Shafer, MDMedscape General Medicine. 2007;9(1):10. ©2007 Medscape Posted 01/16/2007 Case HistoryThe patient is a 45-year-old, African-American female. She is a former intravenous-drug user diagnosed with HIV-1 in 1989. She was first seen at the Stanford Positive Care Clinic (Stanford, California) in 1998 after being treated with zidovudine (ZDV) + didanosine (ddI) from 1996 to 1997, and with ZDV + ddI + lamivudine (3TC) from 1997 to 1998. Her CD4+ cell count at the time was 152 cells/microliter (mcL) and her plasma HIV-1 RNA level was 400,000 copies/mL. She was coinfected with hepatitis C virus subtype 1A. An HIV-1 drug-resistance genotype showed the presence of the uncommon "multinucleoside reverse transcriptase (RT) inhibitor" complex of Q151M-associated mutations, which confers high-level resistance to ZDV, ddI, stavudine (d4T), and abacavir and intermediate resistance to tenofovir, 3TC, and emtricitabine (FTC). In combination with M184V, which confers high-level resistance to 3TC and FTC, the patient's genotype indicated high-level resistance to all of the nucleoside RT inhibitors (NRTIs) available at the time ( Table ). At Stanford, the patient was treated with d4T + ddI + indinavir (IDV) and had a 2.5-log (300-fold) decrease in plasma HIV-1 RNA and an approximately 200 cell/mcL increase in CD4+ cell count. Her plasma HIV-1 RNA level decreased to 600 copies/mL but never reached undetectable levels and rebounded to about 30,000 copies/mL within the year. The sequence of the rebounding virus had the protease inhibitor (PI)-resistance mutations N88S + L90M. Although N88S and L90M are not classic IDV-resistance mutation, they occur, respectively, in about 1% and 15% of viruses obtained from persons with virologic failure who received IDV as their only PI (see PR position summary page). Moreover, each of these mutations appears to cause moderate reductions in IDV susceptibility.[1] Between 1999 and May 2006, the patient was treated with 6 different regimens of highly active antiretroviral therapy (HAART) and experienced transient decreases of plasma HIV-1 RNA of 1 log or more on 3 additional occasions that coincided with the administration of lopinavir/ritonavir (LPV/r) in 2000, tenofovir (TDF) + efavirenz (EFV) in 2001, and enfuvirtide (ENF) in 2003. During this time, CD4+ cell counts remained in the range of 300-500 CD4+ cells/mcL. In June 2004, a liver biopsy, performed because of a decreasing ratio of AST to platelet count, showed stage 4 cirrhosis. Between April 2005 and January 2006, the patient was enrolled in a study for treatment of hepatitis C virus (HCV) and received pegylated interferon alpha-2a with ribavirin. Although her HCV viral load declined, she never achieved clearance. Between mid 2005 and May 2006, the patient's CD4+ cell count decreased from 279 cells/mcL to 132 cells/mcL, reaching her previous nadir. A resistance assay (PhenoSenseGT) showed intermediate genotypic and phenotypic resistance to TDF and high-level genotypic and phenotypic resistance to all other antiretroviral drugs licensed by the US Food and Drug Administration. Present were only 2 of the 21 tipranavir (TPV)-associated mutations reported by Baxter and colleagues[2] and 1 of the 11 darunavir (DRV)-associated mutations reported by De Meyer and coworkers.[3] DRV susceptibility was not performed. The replication capacity (RC) was 3.6%. Because of her declining CD4+ cell count, the patient was enrolled into an open-label trial of a new integrase inhibitor but was randomized to the control arm and was treated with TDF/FTC, ENF, and DRV/r. On this regimen she experienced marked virologic suppression and has had plasma HIV-1 RNA levels below the level of detection beginning in late July and extending until the present time (late November 2006) (Figure). 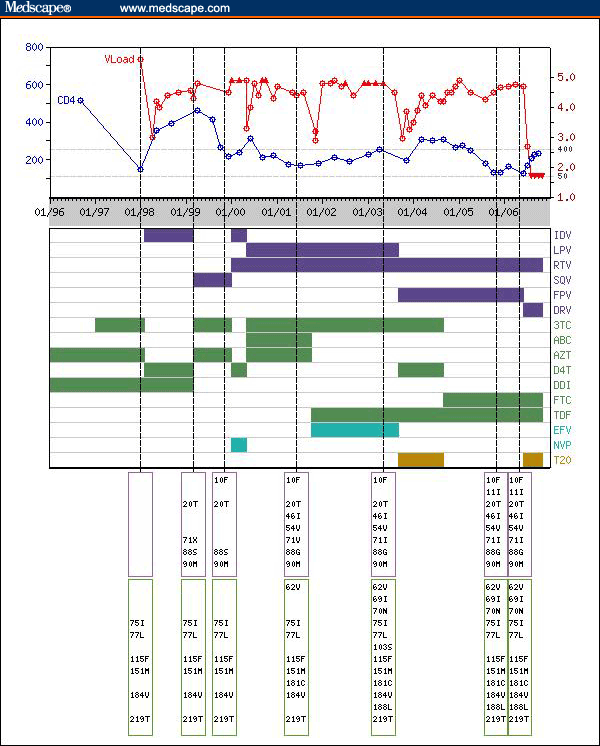 Figure. DiscussionThe first 10 years of this patient's treatment course illustrate the difficulty in treating persons who began their treatment in the pre-HAART era with suboptimal antiretroviral therapy. The subsequent administration of new drugs belonging to new classes often fails in these patients and has been likened to the administration of "sequential monotherapy." The highly successful response to salvage therapy with the use of darunavir/r (DRV/r) and reuse of ENF was not expected. Although sustained plasma HIV-1 RNA response is not yet guaranteed for this patient, this remarkable response in a patient in whom many previous regimens had failed contains lessons about managing treatment-experienced patients with highly drug-resistant HIV. This patient's virus had uncommon mutations in both RT and protease. The Q151M complex of mutations consists of the primary 2-base-pair substitution Q151M in combination with the accessory mutations A62V, V75I, F77L, and F116Y. With the exception of A62V, these accessory mutations do not occur in the absence of Q151M. The complex of mutations causes high-level resistance to ZDV, d4T, ABC, and ddI and intermediate resistance to TDF, 3TC, and emtricitabine (FTC).[4,5] The Q151M complex was most often selected in the pre-HAART era by regimens that contain ZDV or d4T together with ddI. This complex of mutations previously occurred in about 2% of treated persons but has decreased in frequency due to the decline in the use of these nucleoside combinations. The protease sequence from May 2006 contains the PI-resistance mutation L90M, which distorts the substrate cleft indirectly, thereby causing low-level resistance to most PIs. It also contained the 2 common protease flap mutations M46I and I54V. However, it lacks any mutations that are in the substrate cleft in direct contact with PI molecules (eg, mutations at positions 32, 48, 50, 82, or 84). In fact, it is possible that the extraordinary high levels of PI resistance in the May 2006 virus are due in part to mutations that are extremely rare and not well characterized. For example, the effect of N88G on PI susceptibility has not been studied. However, this mutation, which occurs in about 0.2% of viruses from PI-experienced patients, may be important because a similar mutation at this position N88S (both S and G are small amino acids) has a profound effect on several PIs, causing by itself a 20-fold decrease in nelfinavir (NFV) susceptibility, a 10-fold atazanavir (ATV) susceptibility, and a 2- to 5-fold decrease in IDV susceptibility.[1] Atazanavir resistance in a PI-naive patient treated with atazanavir/ritonavir was associated with development of high-level atazanavir resistance and the N88S mutation in protease.[6] Two additional highly unusual mutations were also present in the latest protease sequences, V56A and Y59H. These 2 mutations, which appeared only late in the patient's treatment course, are at highly conserved positions of the protease and have never been studied for their possible effect on PI susceptibility. For HIV-1 isolates with complex genotypes such as those of this patient, susceptibility testing is essential to identify the most active PI. Indeed the patient's virus would have been predicted to be susceptible to TPV/r on the basis of the algorithm developed by Baxter and colleagues because it contained only 1 of the 21 mutations in the TPV/r genotype score. Yet the virus had a 22-fold decrease in TPV susceptibility, which is well above all proposed TPV/r clinical cut-offs.[7] The algorithm for DRV/r may have worked well for this virus as its sequence contained only 1 of the 11 proposed DRV-associated mutations (V11I). DRV susceptibility testing was not performed because the susceptibility assay was performed just prior to DRV approval. The development of genotypic interpretation algorithms for predicting phenotype and virologic response to a new treatment regimen in heavily treated patients is challenging. The first task -- predicting phenotype -- is made more complex by the complicated patterns of mutations that are frequently observed in many heavily treated patients. The second task -- predicting virologic response -- is complicated by the many variables other than the baseline genotype that influence virologic response to a particular drug (eg, previous antiretroviral drugs, baseline virus load, baseline CD4+ cell count, and the coadministered antiretrovirals). The dataset used to develop the TPV/r genotypic interpretation algorithm (baseline genotypes and response to salvage therapy in the RESIST trials) is one of the largest datasets ever used for developing a genotypic interpretation algorithm for a single drug. Moreover, the methods used to develop the TPV/r algorithm[2] are based on a comprehensively documented analysis linking genotype to clinical response. Nonetheless, because of the complexity of patients undergoing deep salvage therapy and the complexity of the genetic mechanism by which TPV resistance develops, further refinements to the algorithm may eventually be proposed. The dataset used to develop the DRV/r genotypic interpretation algorithm (baseline genotypes and response to salvage therapy in the POWER studies) was smaller than that used for TPV/r. Nonetheless, this algorithm is somewhat simpler in that it involves only 11 mutations, most of which also reduce susceptibility to amprenavir, a drug that is structurally similar to DRV. The performance of the TPV/r and DRV/r predictive algorithms has not yet been assessed on independent clinical datasets. When HIV-1 develops many drug-resistance mutations, it often replicates less well than wild-type. Therefore, the extremely low RC of this patient's virus is not surprising. Previous studies have shown an inverse relationship between RC and CD4+ cell count in patients with uncontrolled viremia.[8,9] However, it is not currently known how to exploit the RC assay in order to optimize new treatment regions. Although gp41 sequencing was not done, it is almost certain that this patient's virus developed ENF resistance when virologic rebound occurred in late 2003. ENF has a low genetic barrier to resistance.[10,11] However, ENF mutations are associated with decreased replication,[12] and it is just as likely that the patient's predominant virus population reverted almost entirely to wild-type during the 2 years since ENF was discontinued. Re-treatment with ENF has been described in the DRV registration trials POWER 1 and 2, but there has been no published assessment of the efficacy of ENF re-treatment. In the case of this patient, it is likely that both ENF and DRV/r were responsible for virologic suppression. The maxim that a patient's first regimen has the greatest chance of success remains valid. However, it is still possible to control virus replication in patients in whom multiple previous treatment regimens failed. Such late successes may occur either because of improved adherence or the right combination of newly developed antiretroviral drugs. In the case of this patient, we believe that DRV, which has been highly successful in the POWER trials,[13,14] was the new drug largely responsible for the patient's dramatic response, and that the likely activity of ENF against the vast majority of the patient's actively replicating virus population also played a role. The retained partial activity of the NRTIs in the regimen also may have significantly contributed to her response, as NRTIs retain partial activity even with apparent high levels of resistance. We also anticipate that many patients with highly resistant viruses will achieve similar successes in the upcoming months with the arrival of the investigational integrase inhibitor MK-0508 via expanded access.[15,16] Readers are encouraged to respond to George Lundberg, MD, Editor of MedGenMed, for the editor's eyes only or for possible publication via email: glundberg@medscape.net Table. Summary: PhenoSenseGT Report, May 2006
References
Nancy Shulman, MD,
Assistant Professor of Medicine, Division of Infectious Diseases,
Department of Medicine, Stanford University, Stanford, California;
Division of Infectious Diseases, Palo Alto VA Medical Center, Palo
Alto, California Robert W. Shafer, MD, Associate Professor, Division of Infectious Diseases, Department of Medicine, Stanford University, Stanford, California Disclosure: Nancy Shulman, MD, has disclosed that she has received honoraria and consulting fees from GlaxoSmithKline. Disclosure: Robert W. Shafer, MD, has disclosed that he has received grants for clinical research from Abbott Laboratories, Bristol-Myers Squibb, Celera Diagnostics, Gilead Sciences, GlaxoSmithKline, and Hoffman-LaRoche, and has served as an advisor or consultant for Bayer Diagnostics, Bristol-Myers Squibb, Celera Diagnostics, and Hoffman-LaRoche. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||